
Janis Weis Lab
Scholarly Emphasis: Lyme Arthritis
We study the pathogenesis of Lyme disease; an infection caused by the tick-borne spirochete Borrelia burgdorferi that is responsible for 500,000 infections per year in the US. A spectrum of disease symptoms and severity are observed in infected patients, with arthritis being one of the most common. Using forward genetics we have identified host genes that regulate the severity of Lyme arthritis in inbred strains of mice. Upon infection with B. burgdorferi, C3H mice display severe disease while disease is mild in C57BL/6 mice. We have characterized numerous quantitative trait loci (QTL) that regulate the severity of Lyme arthritis. One of these, Bbaa2, identified the beta-glucuronidase gene (Gusb) as a major regulator of Lyme arthritis severity, with the defective allele resulting in accumulation of pro-arthritic glyclosaminoglycans in the joint tissue of infected mice. Gusb polymorphism also regulates the severity of an autoimmune rheumatoid arthritis model, suggesting variant alleles should be considered risk factors and could serve as targets for treatments for Lyme and rheumatoid arthritis patients.
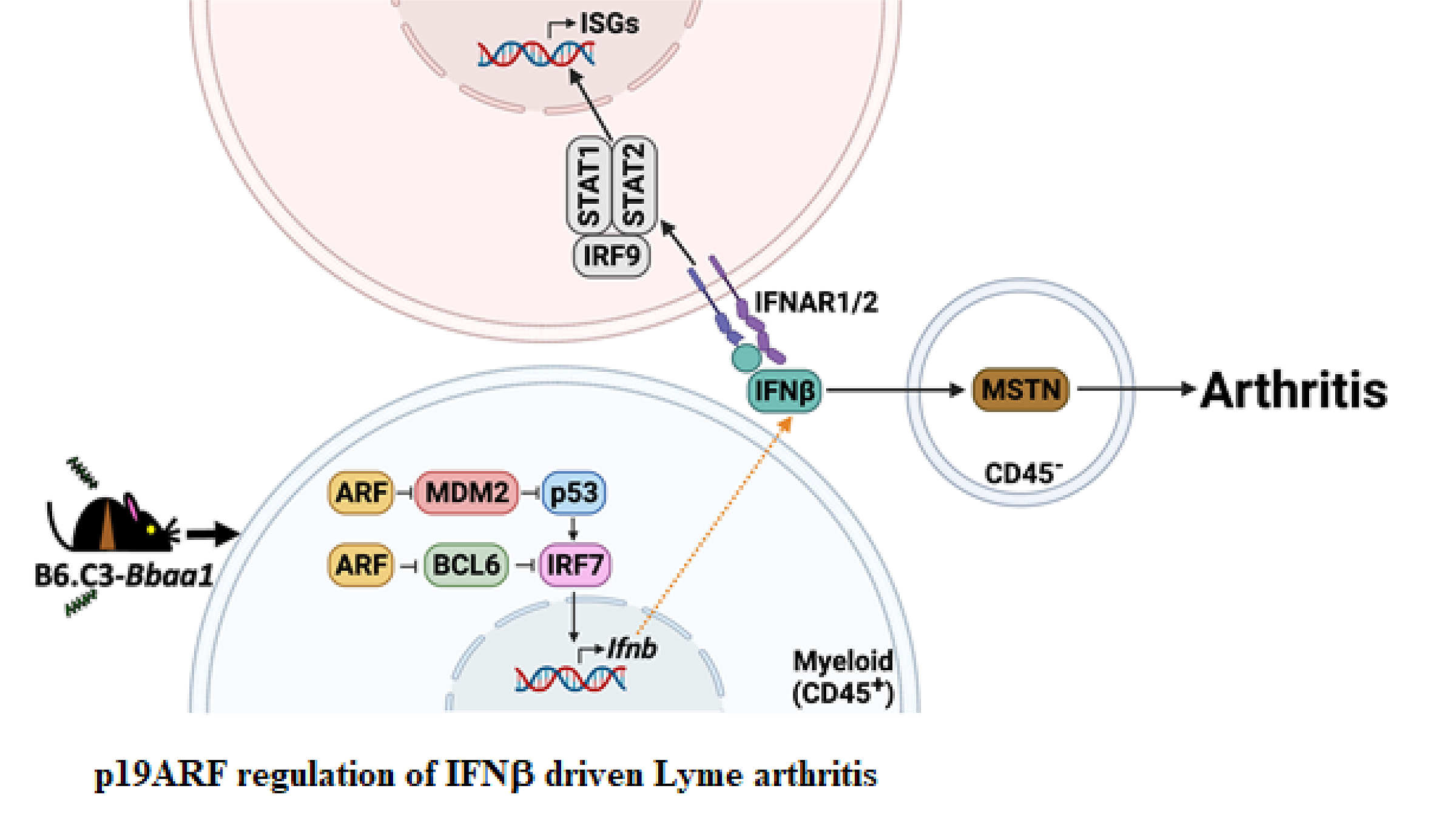
The elevated expression of the C3H allele of p19ARF is responsible for the hyper-induction of IFNb during B. burgdorferi infection of B6.C3-Bbaa1 mice, and resultant greater severity of Lyme arthritis. p19ARF acts to block the ubiquitin ligase MDM2, resulting in stabilized p53 and enhanced expression of IRF7 and IFNb. In a parallel pathway, p19ARF blocks the transcription repressor BCL6, further promoting increased expression of IRF7 and IFNb. Expression of downstream interferon stimulated genes via the STAT1-STAT2-IRF9 signaling complex promotes arthritis and results in regulation of the muscle regulatory protein myostatin, an essential mediator of Lyme arthritis development in B6.C3-Bbaa1 mice. (Figure was created with BioRender.com.)
A second QTL, Bbaa1, includes the Type I interferon (IFN) gene cluster, of particular interest as Type I IFN expression precedes the development of Lyme arthritis in joint tissues of C3H mice and is observed in Lyme disease patients. Positional cloning identified the Cdkn2a gene and its encoded protein, p19ARF, as the Bbaa1 regulator of IFNβ induced by B. burgdorferi. Both siRNA silencing and overexpression of p19ARF confirmed the association. p19ARF was found to act through parallel pathways involving the stabilization of the tumor suppressor p53 and sequestration of the transcriptional repressor BCL6. Our findings linking p19ARF regulation of p53 and BCL6 to the severity of IFNβ-induced Lyme arthritis in vivo reveal potential roles for p19ARF, p53, and BCL6 in Lyme disease and other IFN hyperproduction syndromes. Our studies further identified the muscle regulatory gene myostatin as a critical downstream mediator of Lyme arthritis development in mice, and point to additional therapeutic targets in arthritis development.

Borrelia burgdorferi–infected mice expressing a single TCR transgene for a non–Borrelia epitope develop severe Lyme arthritis, revealing bystander activation of T cells is sufficient to promote disease. J. Immunol. 200: 1457–1470.
We have also developed a model for one of the most troubling aspects of Lyme disease, the persistence of symptoms after what should have been an effective antibiotic regimen: post treatment Lyme disease syndrome (PTLDS). We have discovered that arthritis is sustained by bystander activation of CD4+ and CD8+ T lymphocytes. These T cells become a source of arthritis promoting IFNg, leading to inflammatory markers with striking similarity to those seen PTLDS patients. Bystander activated CD4+ and CD8+ bystander T cells from B. burgdorferi-infected mice display upregulated TLR2, which is central to their activation and promotion of arthritis. We are working to characterize bystander-activated T cells through single cell expression profiling, with the goal of identifying unique T cell markers as potential targets for resolution of arthritis.
Two additional Lyme arthritis models that exploit a dysregulated innate host response have been identified in collaboration with Ryan O’Connell’s lab: the miR146a-/- mouse and the miR-155-/- mouse. Previous studies had revealed the central role of the TLR2 adapter MyD88 and the NF-kB dependent transcriptional response in control of B. burgdorferi infection. Studies with miR146a-/- and miR-155-/- mice demonstrate that NF-kB dependent cytokines are also critical for the inflammatory responses leading to arthritis and carditis. These mice will provide models for testing novel therapeutics that reduce inflammation without impairing host defense.






Publications
Lab Twitter Feed
Current Funding
Borrelia burgdorferi Mitogen in Development of Arthritis, 5R01AI032223-24
A small percentage of patients infected with the Lyme disease pathogen Borrelia burgdorferi develop symptoms that persist following antibiotic treatment, termed post treatment Lyme disease syndrome, PTLDS. Studies on the underlying mechanism of PTLDS have been hampered by lack of an animal model. We have recently developed a mouse model of PTLDS and found that bystander activated T cells drive sustained symptoms of disease and display a novel gene expression signature. T cell intrinsic expression of TLR2 and other pathogen receptors are hypothesized to circumvent the normal strict tolerance of antigen specific T cell activation, and are proposed to provide novel targets for re-setting the inflammatory response in PTLDS patients. We have further confirmed the presence of bystander activated T cells in the peripheral blood of patients with PTLDS, validating the appropriateness of the mouse model and its potential utility in testing novel therapeutics.
Molecular Genetics of Lyme Arthritis, 2R01AR043521-26
Lyme disease, caused by the spirochete Borrelia burgdorferi, is associated with a spectrum of clinical symptoms and a dramatic range in severity among infected individuals. A forward genetics approach identified a locus on Chr 4 of the mouse that regulates Lyme arthritis by controlling the expression of Type I IFN, and downstream induction of MSTN as an effector of arthritis development. The mechanism by which Type I IFN and MSTN contribute to Lyme arthritis development will be addressed, with the goal of identifying novel therapeutic targets for patients with severe disease.